
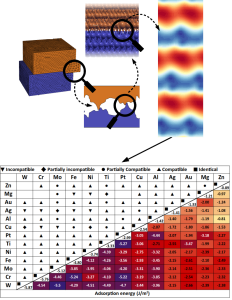
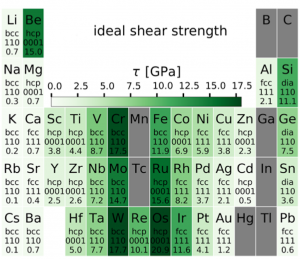
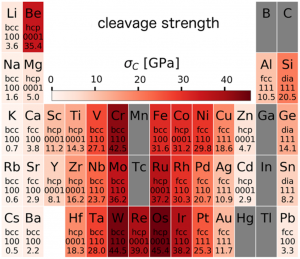
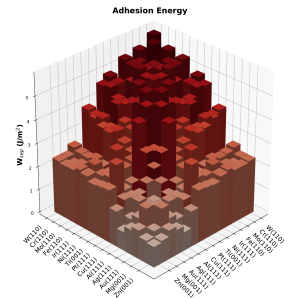
The use of first-principles calculations based on density functional theory (DFT) to screen the properties of many materials in parallel and in an automatized way represents an exciting opportunity for discovering new materials and/or general trends.
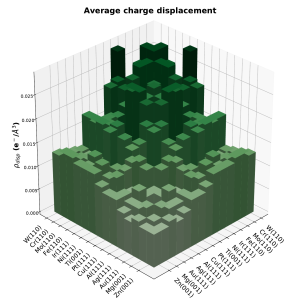
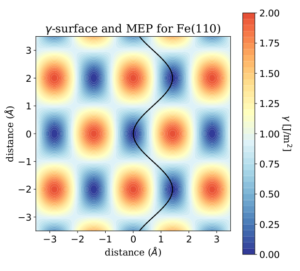
We developed workflows, XSORB and Tribchem, for the automatized screening of solid surfaces and interfaces. The calculated properties include surface adsorption, interfacial adhesion, charge displacements, and ideal shear strength.[47, 53] The latter is obtained from the first-principles calculated potential energy surface (PES) (or Generalized Stacking Fault Energy Surface) according to the procedure developed by our group [23].
Through this approach we calculated the adhesion energies of several metallic interfaces [82], and analyzed how chemical surface modifications affect them [96]. Metal-semiconductor [101], 2D [34,74], 2D-metal [77] and other classes of solid interfaces relevant to technological applications are under investigation.
The databases we are continuously populating are available here.
Monitoring system evolution through molecular dynamics and multiscale schemes
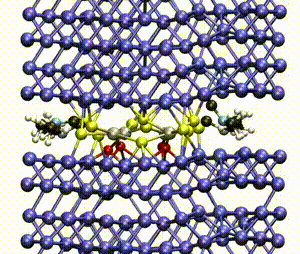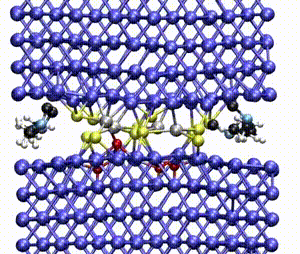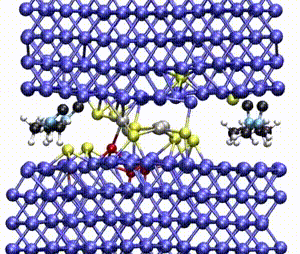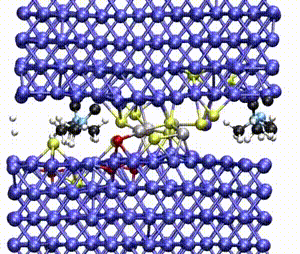
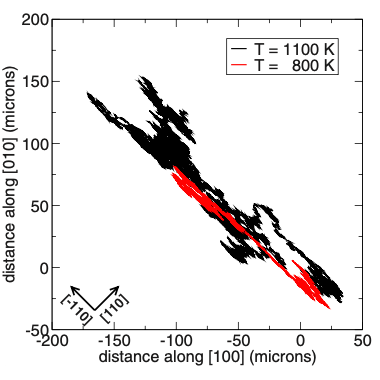
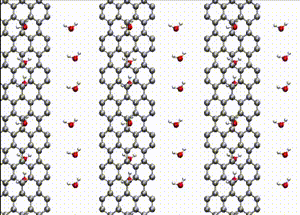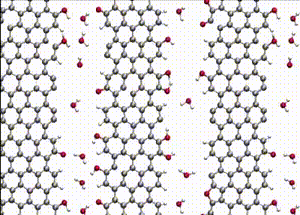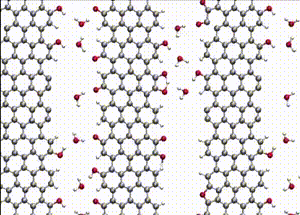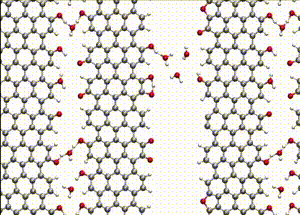
Computer simulations offer a unique opportunity to open a window onto the complex scenario of the atomistic events occurring during system evolution. This is highly relevant for understanding and predicting the material response to external stimuli, such as temperature and mechanical stress.
We apply atomistic simulations to monitor, in real time, processes such as tribochemical and catalytic reactions, which require an accurate description of material chemistry. Starting from a brute-force use of ab initio molecular dynamics (AIMD), as in e.g., [30, 48, 88, 89], or hybrid schemes such as QM/MM [57, 59, 61] and kinetic Monte Carlo [6, 9, 11], we have now moved to mD based on machine learning potentials (MLPs).
Thanks to the SCS software, we have automatized the active learning training of MLPs, able to accurately describe the atomic interactions even in extreme conditions, and significantly increased the time and length scales of our simulations [98, 100, 107, 109].
We have also developed a multiscale scheme that links AIMD to Green’s functions MD for both the accurate description of interfacial chemistry and the elastic response of semi-infinite bulks [86].