


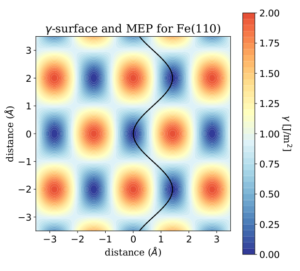








Due to the wide number of possible homogeneous and heterogeneous solid interfaces of interest for tribology, it is convenient to employ automatic workflows able to screen the physical properties of all these materials. Among the most relevant properties, the potential energy surface (PES), the charge displacement, and the shear strength provide essential insights on the tribological behavior of the different interfaces. Automatic workflows, like the one implemented in our group, are capable to calculate the most important figures of merit of hundreds of different solid-solid interfaces [1,2].
The lubricant materials currently employed to reduce friction can be divided in solid lubricants, active in dry conditions, and lubricant additives, included in a liquid medium. In our group we study by means of first principles simulations the tribological properties of these materials and their interactions with the substrates. First principles simulations are particularly suited for this task because they can accurately describe all the degrees that are necessary to correctly model tribochemical reactions, i.e. the chemical reactions occurring in the presence of mechanical stresses. We study carbon-based materials [1,2,3], layered materials such as phosphorene and molybdenum disulfide [3,4], extreme pressure additives such as MoDTC and ZDDP [5,6,7] and the effect of passivating species on the sliding surfaces [8,9].
To reduce the computational effort associated to fully ab initio molecular dynamics, which can accurately describe tribochemical reactions, hybrid schemes can be adopted. The quantum mechanics/molecular mechanics (QM/MM) approach we have recently adopted allows
to focus the computational power on the portion of the system where the chemical reactions occur and
where the high accuracy is needed, without spending unnecessary resources on the portion of the system where the details of the electronic structure are not needed. This approach has been proven successful to describe the tribochemistry of water at the graphene edges [1,2] and MoDTC additives [3].
Furthermore, high forces must be dissipated during sliding. This is most often achieved by phonons. Green’s functions molecular dynamics (GFMD) simulations and the relevant theory have been proven successful to overcome the limitation of finite-size simulations system, in which phononic dissipation cannot be easily represented. Our GFMD method can be applied also to large-scale simulations of systems that are impossible to calculate due to the computational cost.
© 2026 Computational materials & tribology. Built using WordPress and the Mesmerize Theme
| Cookie | Duration | Description |
|---|---|---|
| cookielawinfo-checbox-analytics | 11 months | This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics". |
| cookielawinfo-checbox-functional | 11 months | The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional". |
| cookielawinfo-checbox-others | 11 months | This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other. |
| cookielawinfo-checkbox-necessary | 11 months | This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary". |
| cookielawinfo-checkbox-performance | 11 months | This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance". |
| viewed_cookie_policy | 11 months | The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data. |